Guide
bioimageit_core is a python3 library. It implements the main API for BioImageIT middleware.
In the scheme below we can see the position of bioimageit_core in the BioImageIT ecosystem. In fact, bioimageit_core is the
API that connects low level image processing and data management to high level end users applications.
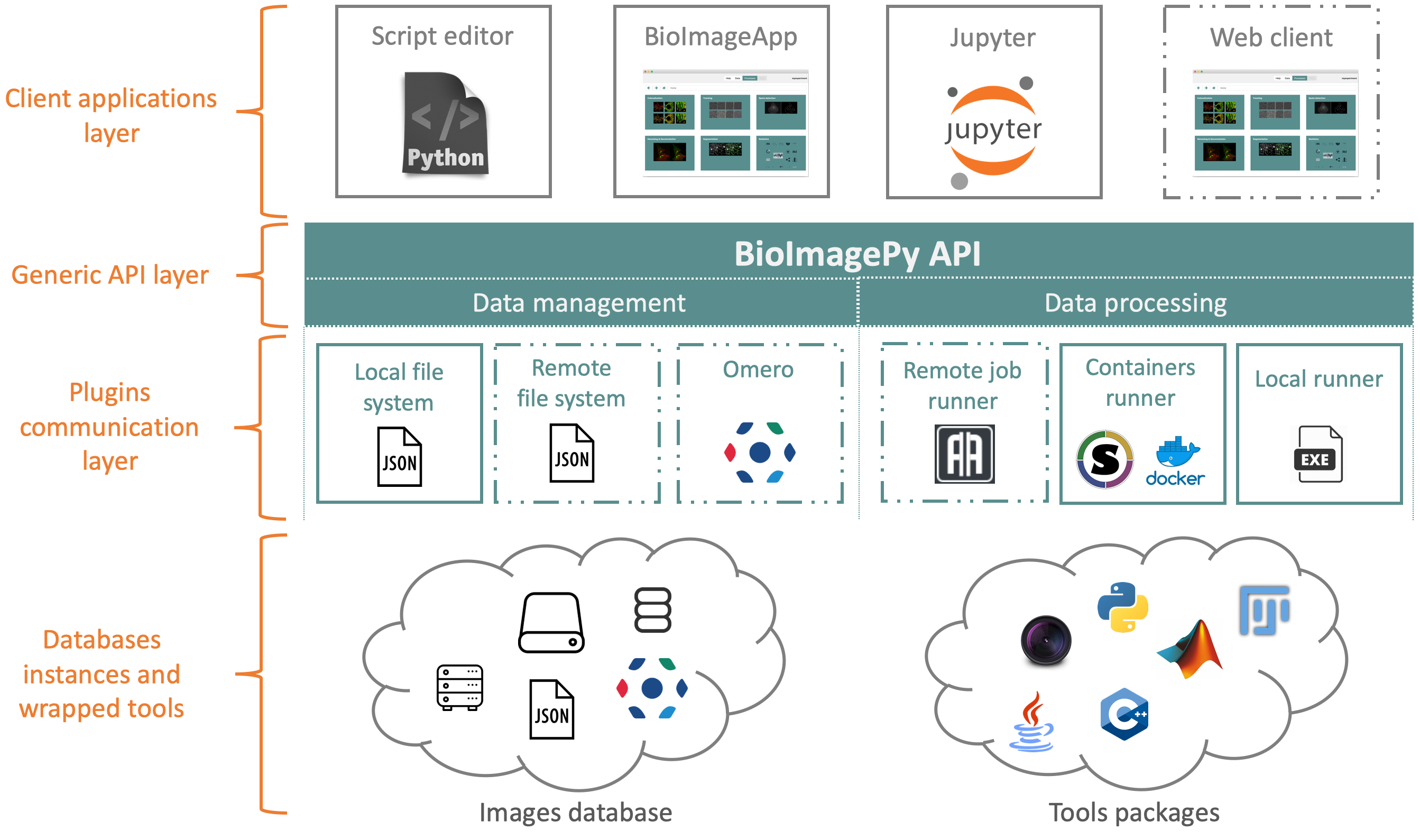
For data management, bioimageit_core implements a set of functions to manage and annotate data at the Experiment (ie project)
level. For image processing tools, bioimageit_core implements a set of functions to query a tool database and
to run tools on data.
For Data management and tools management, bioimageit_core defines an API that can be implemented with plugins. By
default, data is managed using JSON files locally. If we want to use the bioimageit_core API to manage data on
a SQL database for example, we can implement a data management plugin that links the bioimageit_core API with the SQL database.
For tools execution, bioimageit_core by default runs tools using Conda packages in the local machine. If we want to run a processing
tool with a job scheduler for example, we can write a tool runner plugin that links the bioimageit_core API to the job scheduler.
The advantage of this bioimageit_core architecture is to enable writing high level python code to manage and annotate data and deploy it
in different hardware or network architecture without the need to update the high level code. Only plugins have to be added.
Data Management
In the BioImageIT project, we propose to manage data using a 3 layers representation:
Experiment: an experiment is a project that contains one dataset of raw data named “data” and a list of processed datasets. Each processed dataset contains the outputs of a processing tool.
DataSet: a dataset contains a list of data that can be raw or processed
Data: a data contains a single data and the associated metadata. For a
RawDatametadata are a set of key:values pairs information to identify data and a generic dictionary for any specific metadata (like image resolution…). ForProcessedData, metadata are a link to the origin data and the run information.
In this section we show the main functions implemented in the bioimageit_core library to handle Data, Dataset and Experiment. Please
refer to the docstring documentation to get more advanced features.
The BioImageIT API is accessible through a single class named Request.
import bioimageit_core.api as iit
req = iit.Request('./config_sample.json')
req.connect()
To create an experiment, bioimageit_core has a dedicated function with the following syntax:
experiment = req.create_experiment(name='myexperiment',
author='Sylvain Prigent',
date='now',
destination="./")
This creates an empty project with the basic metadata of the experiment. Then we can import a single data:
req.import_data(experiment,
data_path='data_uri',
name='mydata',
author='Sylvain Prigent',
format_='imagetiff',
date='now',
key_value_pairs={"key": value})
or a multiple data from a directory:
req.import_dir(experiment=experiment,
dir_uri='./tests/test_images/data',
filter_=r'\.tif$',
author='Sylvain Prigent',
format_='imagetiff',
date='now')
Then the next step is to annotate the data. It can be done in batch with functions like:
req.annotate_from_name(experiment, 'Population', ['population1', 'population2'])
or:
req.annotate_using_separator(experiment, 'ID', '_', 1)
that will create key-value pairs for each data by extracting information from the data file names. The first case will search the words population1 and population2 in the data file name and associate it to the key population if one of the the words population1 or population2 is found. The second case shows how to extract information from the data file name using separators. Here we extract the sub-string in the file name that is located between two _ and after the first _, and associate the extracted value to the key ID. We can also manually annotate one data by extracting it and manually adding a key-value pair:
data = req.query(experiment, dataset_name='data', query='name=population1_001.tif')
data[0].set_key_value_pair("population", "Population1")
data[0].set_key_value_pair("ID", "001")
req.update_raw_data(data[0])
The bioimageit_core library also allows to access directly a DataSet:
raw_dataset = req.get_dataset(experiment, name='data')
and interact with the data in the DataSet:
data = req.get_data(raw_dataset, query='Population=population1')
Process Running
In the BioImageIT project processing tools are external packages (like Conda packages or Docker containers) and represented with XML wrappers similarly to the Galaxy Project.
The bioimageit_core library, implements functionalities to manipulate and run packaged tools.
A Tool is a python class in bioimageit_core that allows to identify a processing tool. It load the tool XML file and allows to print and access the process information.
tool = req.get_tool('spitfiredeconv2d_v0.1.2')
tool.man()
We can see that we do not need to instantiate directly a Tool since bioimageit_core manage a
process database. We can then access a Tool simply using the tool name and version:
A tool can be ran in data file directly with the exec command:
req.exec(tool,
i='tests/test_images/data/population1_001.tif',
o='population1_001_deconv.tif',
sigma=4,
regularization=12,
weighting=0.1,
method='SV',
padding=True)
Nevertheless, it is not the recommended methods since exec does not generate any metadata. We prefer
using the run method which runs a Job on an Experiment and keep track of all the job history (inputs data, outputs data, parameters).
from bioimageit_core.containers.runners_containers import Job
job = Job()
job.set_experiment(experiment)
job.set_tool(req.get_tool('spitfiredeconv2d_v0.1.2'))
job.set_input(name='i', dataset='data', query='')
job.set_param('sigma', '4')
job.set_param('regularization', '12')
job.set_param('weighting', '0.1')
job.set_param('method', 'SV')
job.set_param('padding', 'True')
job.set_output_dataset_name('deconv')
experiment = req.run(job)
Then, all the output data and the run metadata are stored in a new dataset of the Experiment. In the example above the new dataset is called deconv.
Further reading
In this short introduction guide we show the basic information we need to use bioimageit_core library.
For a more advanced use, we recommend reading the following tutorials.